|
|
Abstract
Objective:
To investigate The level of serum amyloid A
protein (SAA) in systemic lupus erythematosus (SLE) patients and its
importance in disease activity.
Materials and Methods: Forty- two female patients who satisfied
four or more of the revised ACR criteria for SLE were included in this study.
Fifteen healthy female subjects matched for age were included as controls.
Disease activity was assessed by the systemic lupus activity measurement (SLAM)
index. Serum amyloid A protein was measured by the particle enhanced
nephelometry technique.
Results: Serum amyloid A protein (SAA) levels in SLE patients (95.27±100.62
mg/l) were higher than in the controls (4.08+1.14 mg/l) and the difference was
statistically significant. Correlation between the SAA level and some of the
disease parameters revealed a statistically significant positive correlation
with the SLAM index score, prednisone dose and C-reactive protein. No
significant correlation was found with age, disease duration, C3, C4, total
leucocytic count, ESR or hemoglobin. Serum AA was significantly higher among
patients with lupus nephritis
Conclusion: Raised levels of SAA may indicate disease activity as well
as lupus nephritis in SLE patients but can’t be used for monitoring of responses
in patients receiving systemic corticosteroid therapy.
Introduction
Serum amyloid A (SAA), is a putative precursor molecule of amyloid-A (AA)
protein, formed by proteolytic cleavage by macrophage or polymorph proteinases[1].
It acts as an acute phase reactant, its concentration increases by up to
1000-fold during inflammation, largely owing to cytokine-driven transcriptional
upregulation[2].
SAA can be used as a marker of inflammation in some autoimmune disorders such
as rheumatoid arthritis, and vasculitis [3] It has a number of
immunomodulatory roles. It can induce chemotaxis and adhesion molecule
expression and has cytokine-like properties. SAA was found to promote the
upregulation of metalloproteinases [4] and to induce
collagenase production, providing a means of remodeling the extracellular matrix
in areas of inflammation [5].
SAA transiently binds high density lipoproteins-3- (HDL3)to macrophages
during an inflammatory response. In this way, it can mediate the delivery of
lipids to sites of injury for use in tissue regeneration. However, in chronic
inflammatory conditions, persistently high levels of SAA may compromise normal
cholesterol transport and contribute to the development of atherosclerosis [6].
Binding sites on the SAA protein for calcium, laminin, and heparin
/heparan-sulfate are described as well indicating its ability to affect cell
adhesion, migration, proliferation and aggregation[7]. SAA is
therefore involved in various physiological and pathological processes,
including inflammation, atherosclerosis and thrombosis [8].
The role of elevated levels of SAA over time in predisposision to secondary
amyloidosis is debatable[9]. Acute phase serum amyloid A has
been reported to be more sensitive than C-reactive protein (CRP) as a marker of
disease activity in rheumatoid arthritis [10]. In this study
the profile of SAA in systemic lupus erythematosus patients was studied in
relation to clinical manifestations and disease activity.
Methods
A. Patients:
Forty two patients with systemic lupus erythematosus (SLE), attending the
Rheumatology and Rehabilitation, internal medicine and dermatology outpatient
clinics of Cairo University Hospitals were included in this work. Their ages
ranged from 20 to 47 years and the disease duration from 0.25 to 15 years. All
patients satisfied 4 or more of the revised American College of Rheumatology (ACR)
criteria for classification of SLE [11, 12].
Fifteen healthy female subjects with matched ages served as controls.
B. Clinical assessment:
Full history taking, clinical examination and laboratory investigations were
tabulated in accordance to the systemic lupus activity measurement (SLAM) index
[13]. This index covers the symptoms that occurred over the
previous month and includes 24 clinical and 8 laboratory variables. In addition
serum ANA, anti-n-DNA, serum complement “C3 and C4” and C-reactive protein were
also assessed.
Patients with obvious bacterial infections and impaired renal functions were
excluded as serum amyloid A was reported to increase in these conditions [14],
[15].
C. Assessment of serum amyloid A:
- Collection of samples: 5 ml venous samples were withdrawn from patients and
controls and stored frozen at –200C.
- Determination of SAA was done by the particle-enhanced immunonephelometry
method [16], on the Behring Nephelometer (BN II), Dade Behring
Inc., N.Y., U.S.A.
Statistical Methods: Data were processed on a personal computer utilizing the SPSS® 13.0 for Windows® statistical package. Student’s t-test was used when
appropriate. Two-tailed analysis with P value less than 0.05 was considered
significant. Range, mean, and standard deviations are given. Correlation
analysis was performed using Pearson’s correlation.
Results
The general characteristics and clinical features of the studied group are
shown in table (1).
Thirty four (80.95%) of the patients were receiving oral steroid therapy. The
dose ranged between 5 - 60 mg daily, with a mean of 18.9±12.6 mg /day, for at
least 6 months before the time of study. Eight (19.05%) cases were new and
hadn’t received any treatment yet.
The serum amyloid A level of SLE patients ranged from 2.6- 532 mg/l with a mean
of 95.27±100.62 mg/l, while those of the control group ranged from 2.8-5.9 mg/l
with a mean of 4.08±1.14; the difference was statistically significant
(P=0.0001), table (2).
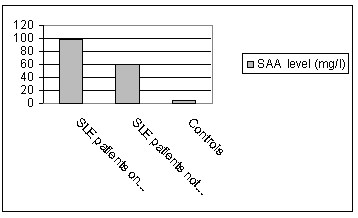
Serum amyloid level in patients who hasn’t received any treatment (mean=
60.34+99.35) were studied. It was significantly higher than controls (P= 0.043)
but it was lower than in cases under steroid therapy (regardless their response
to treatment) (98.29+ 100.74) and the difference was statistically significant
(p=0.039) figure (1).
Correlation between the serum amyloid A level and some of the disease parameters
revealed statistically significant positive correlation for the SLAM index score
(r=0.377, P=0.02), prednisone dose (in cases under treatment) (r=0.344, P=0.04)
and C-reactive protein (r=0.698, P=0.0001) table (3).
Serum amyloid A levels were compared in SLE patients with and without some
clinical and laboratory parameters of the disease. The mean serum amyloid A was
significantly higher among patients with nephritis (P=0.03), table (4).
|
Table (1): General characteristics and
clinical features of SLE patients: |
| Criteria |
SLE patients (N = 42) |
Age (years)
Age of onset (years)
Disease duration (years)
|
25.41±7.32
21.39±5.86
4.31±3.11
|
Constitutional:
Fever
Fatigue
Weight loss
|
25 (59.52%)
31 (73.81%)
29 (69.05%)
|
Mucocutaneous:
Oral ulcers
Malar rash / photosensitivity
Discoid LE lesions
Alopecia:
Cicatricial
Diffuse noncicatricial
Vasculitis:
Hands:
Palmar erythema
Erythema multiforme like lesions
|
25 (59.52%)
34 (80.95%)
4 (9.52%)
26 (61.90%)
3
23
8 (19.05%)
34 (80.95%)
34 (80.95%)
21 (50%)
|
Joint:
Arthralgia
Arthritis
|
38 (90.05%)
21 (50%)
|
|
Pulmonary |
16 (38.1%) |
|
Raynaud’s phenomenon |
14 (33.33%) |
|
Carditis |
7 (16.67%) |
|
Hypertension |
22 (52.38%) |
|
Nephritis |
17 (40.48%) |
|
Myalgia/Myositis |
8 (19.05%) |
|
CNS affection |
19 (45.24%) |
Hematological:
Leucopenia
Thrombocytopenia
Hemolytic anemia
|
9 (21.43%)
5 (11.9%)
-
|
Autoantibodies:
ANA
Anti-n-DNA
|
40 (95.24%)
28 (66.67%)
|
|
Table (2): Comparison between mean serum amyloid-A
level of SLE patients and control. |
|
|
SLE patients
N= 42 |
Control group
N= 15 |
P |
|
Mean±SD |
95.27±100.62 |
4.08±1.14 |
0.0001* |
|
* statistically significan P< 0.05 |
|
Table (3): Correlation between serum amyloid-A and
some of the disease parameters in SLE patients. |
|
|
r |
P |
|
Age |
-0.221 |
0.208 |
|
Disease duration |
-0.093 |
0.601 |
|
SLAM score |
0.377 |
0.02* |
|
ESR |
0.297 |
0.088 |
|
HB |
-0.273 |
0.118 |
|
TLC |
-0.008 |
0.966 |
|
C3 |
-0.226 |
0.2 |
|
C4 |
-0.043 |
0.809 |
|
C-reactive protein |
0.698 |
0.0001* |
|
Prednisone dose |
0.344 |
0.04 |
|
* statistically significant P< 0.05. ESR=Erythrocyte
sedimentation rate,
r=
Correlation
coefficient, HB= Hemoglobin, TLC=Total leucocytic count |
|
Table (4): Comparison of
mean serum amyloid-A according to the presence of some disease parameters
in SLE patients. |
|
|
Present |
Absent |
P value |
|
Fever |
104.84±117.8 |
86.28±60.93 |
0.615 |
|
Oral ulcers |
114.27±120.09 |
69.0±38.3 |
0.215 |
|
Raynaud’s |
105.89±148.02 |
94.65±72.26 |
0.766 |
|
Arthritis |
104.75±131.13 |
91.02±52.28 |
0.698 |
|
Nephritis |
138.18±128.08 |
66.79±58.94 |
0.03* |
|
Serositis |
106.52±78.39 |
91.79±117.16 |
0.679 |
|
CNS |
91.28±74.39 |
104.52±121.37 |
0.708 |
|
Anti-n-DNA |
79.39±73.28 |
125.29±128.76 |
0.195 |
|
Discoid LE |
106.54±64.67 |
117.35±101.55 |
0.164 |
|
Malar rash |
82.63± 68.43 |
105.23±97.36 |
0.561 |
|
* statistically significant P< 0.05 |
|
Figure 1: Effect of systemic
steroids on SAA levels in SLE patients:
|
Discussion
The mean serum amyloid A in systemic lupus erythematosus
patients was significantly higher than the control group. This suggests that SAA
might be related to the disease process in SLE. Other workers have reported that
SAA levels are greatly elevated in rheumatoid arthritis [8,
17]and correlated significantly with disease activity. In SLE some authors
reported high SAA levels in patients with active disease but lower than those
seen in RA patients [18]. Others found that SAA levels in
patients with SLE were only modestly raised, even in those with severe active
disease, unless significant intercurrent microbial infection was also present [19].
However SLE patients with obvious infections were not included in the current
study.
When SAA levels where studied in relation to disease activity parameters in
the studied group, SAA showed a significant positive correlation with the SLAM
index score, C-reactive protein and insignificantly correlated to the ESR. This
significant correlation indicates that disease activity in SLE is associated
with raised SAA levels. This finding could suggest that SAA may be useful as a
disease activity marker in patients with SLE.
A strong correlation between SAA and C-reactive protein in patients with SLE and
other dermatoses was reported by other authors [20]. SAA and
C-reactive protein are under the influence of different cytokines; CRP is
predominantly stimulated by interleukin-6 (IL-6) [21], while
SAA responds preferentially to IL-1 [22], but requires the
synergistic action of both cytokines for maximal stimulation [23].
A selective response in the liver of differing degrees of synthesis and
degradation may favor one acute phase protein over another in different
pathophysiological circumstances [24].
The failure to detect a significant correlation between the SAA level and the
ESR may be attributed to the fact that ESR is only an indirect measure of
disease activity and reflects mainly fibrinogen levels. Furthermore, changes in
the ESR occur slowly and are influenced by factors such as anemia, the size and
shape of red blood cells, lipid levels, and hypergammaglobulinemia [8].
In this study no correlation could be detected between SAA levels and the
presence of arthritis. When compared to ESR or C-reactive protein SAA is
considered the best marker available for the assessment of inflammatory joint
disease in rheumatoid arthritis and ankylosing spondylitis [4,
8, 25]. However according to our findings, this seems not
to be the case for joint inflammation associated with SLE.
Another interesting point of view is the described role of plasma SAA as a
precursor of Amyloid A (AA) protein in secondary amyloidosis an exceptionally
rare complication in SLE [19]. The acute phase response
involves a major rearrangement of plasma protein synthesis by the liver through
increased production of some proteins and reduced levels of others [24].
It is thought that the function of this reaction is to confine the source of
inflammation and limit autolytic damage by phagocytic cells [26].
However, in chronic disease, the continued presence of these proteins may
exacerbate the inflammatory process, directly result in deposition of amyloid
fibrils that contribute to tissue damage [18]. SAA is a serum
precusor of amyloid A protein, the fibrillar component in reactive amyloid
deposits [27].
Compared to rheumatoid
arthritis, secondary amyloidosis is considered a rare complication of SLE
[19]. However in one study it was detected in 7% of SLE
patients namely, those with long standing disease or those with long course of
immunosuppresive therapy [28].
High levels of SAA were found to correspond with the incidence of reactive
systemic amyloidosis in SLE and other inflammatory diseases [19].
Autoantibodies to amyloid A protein were demonstrated in one third of SLE cases
but their presence was not significantly associated with the development of
secondary amyloidosis [29]. Recent studies showed that high
concentration of SAA is not sufficient for the development of amyloidosis and
that genetic susceptibility through polymorphism of the SAA gene is an important
back ground of amyloidogenesis [30, 31].
Those susceptible patients with high risk alleles (SAA 1.5) may be liable to
develop reactive amyloidosis [32].
The present study implied a positively significant correlation of SAA levels
in SLE patients under treatment, with the dose of prednisone they were
receiving. This might be due to the fact that more severe cases required higher
doses of prednisone. On the other hand corticosteroid hormones including
dexamethasone, corticosterone, hydrocortisone, and aldosterone may be involved
in the upregulation of SAA mRNA expression and thereby increase SAA production [33].
They may affect the synthesis of this protein through altering the production of
several cytokines as IL-1, IL-6 [34, 35].
Therefore if elevation of SAA levels is implicated in in the development of
amyloidosis, the propriety of using corticosteroid treatment to the patients at
risk should be considered [31].
In this study patients who hasn’t received treatment had significantly higher
levels of SAA compared to normal controls. These levels were signifcantly lower
when compared to those who received treatment regardless their response to
treatment. This would support the explanation of the positive correlation
between the SAA level and prednisone dose received, is secondary to the
stimulatory effect of systemic steroid therapy on SAA. This finiding may
partially negate its value in monitoring the response to treatment.
In lupus, profound activation of cytokine production and the acute phase
response have been reported to be associated with a markedly increased risk for
the development of atherosclerosis[36]. Moreover, extrahepatic
production of the SAA (SAA1 and SAA2 protein isoforms) in a number of
atherosclerotic lesions including endothelial cells, cultured smooth muscle
cells and monocyte-macrophage cell lines has been reported [33].
In a previous study, women with longer duration and a higher cumulative dose of
of prednisone use as well as those with prior coronary events were more likely
to have carotid atheromatous plaques [37]. High levels of SAA
in cases receiving high doses of systemic corticosteroid may be behind the high
incidence of atherosclerosis in those cases [33].
As regards to the clinical manifestations, in our study the mean SAA level
was significantly higher among patients with lupus nephritis. A significant
relationship between elevated levels of SAA and renal disease was reported [38]
to be due to the presence of inflammation, as evidenced by increased levels of
specific cytokines [35].
Conclusions:
We could therefore conclude that elevated SAA level is a marker that reflects
disease activity in SLE patients especially in cases with nephritis. Since SAA
level correlates positively with corticosteroid doses received, it cannot be
used for monitoring the response of treatment in patients receiving this
medication.
References
1. Cunnane G& Whitehead AS, Amyloid precursors and amyloidosis in rheumatoid arthritis. Baillieres Best Pract.Res.Clin.Rheumatol.13(4):615-28, 1999.
2. Rygg M, Uhlar C.M, Thorn C, Jensen LE, Gaughan DJ, Varley AW, Munford RS, Goke R, Chen Y & Whitehead AS, In vitro evaluation of an enhanced human serum amyloid A (SAA2) promoter-regulated soluble TNF receptor fusion protein for anti-inflammatory gene therapy, Scand J Immunol.53(6):588-95, 2001.
3. Jovanovic DB, Clinical importance of determination of serum amyloid A, Srp Arh Celok Lek.132(7-8):267-71, 2004.
4. O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis, Arthritis Rheum.50(6):1788-99, 2004..
5. Fitzpatrick D, Fisher H. Histamine synthesis, imidazole dipeptides and wound healing. Surgery 1982; 91:430-4.-8.
6. Salazar Soler A, Pinto Sala X, Mana Rey J, Pujol Farriols R, Inflammatory response, cholesterol metabolism, and arteriosclerosis, An Med Interna.18(2):100-4, 2001.
7. Urieli-Shoval S, Linke RP, Matzner Y, Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states, Curr Opin Hematol.7(1):64-9, 2000.
8. Cunnane G, Grehan S, Geoghegan S, McCormack C, Shields D, Whitehead AS, Bresnihan B, Fitzgerald O, Serum amyloid A in the assessment of early inflammatory arthritis. J Rheumatol. 27(1):58-63, 2000.
9. Obata T, Takahashi H, Nosho K, Ikeda Y, Tokuno T, Kawahito Y, Honda S, Makiguchi Y, Imai K, Ikeda T, A case of systemic lupus erythematosus overlapping with progressive systemic sclerosis accompanied by deposition of AA amyloid in the stomach, Ryumachi. 38(6):810-7, 1998.
10. Hilliquin P. Biological markers in inflammatory rheumatic diseases. Cell Mol Biol. 41(8):993-1006, 1995.
11. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield N, Schaller JG, Talal N, Winchester RJ, The 1982 revised criteria for the classification of SLE, Arthritis Rheum. 25: 1271-7, 1982.
12. Hochberg MC, Updating American College of Rheumatology revised criteria for the classification of SLE, Arthritis Rheum. 40:1725, 1997.
13. Liang MH, Socher SA, Larsen MG, Schur PH: Reliability and validity of six systems for the clinical assessment of disease activity in SLE, Arthritis Rheum. 32:1107, 1989.
14. Smith JW, Colombo JL, McDonald TL, Comparison of serum amyloid A and C-reactive protein as indicators of lung inflammation in corticosteroid treated and non-corticosteroid treated cystic fibrosis patients, J. Clin. Lab. Anal. 6 : 219-24, 1992.
15. Hartmann A., Eide T.C. & Fauchald P, Serum amyloid A protein is a clinically useful indicator of acute renal allograft rejection. Nephrol. Dial. Transport. 12 : 161-6, 1997.
16. Malle E, De Beer FC : Human serum amyloid A (SAA) protein : a prominent acute-phase reactant for clinical practice. Eur. J. Clin. Invest. 26 : 427-35, 1996.
17. Benson MD, Cohen AS, Serum amyloid A protein in amyloidosis, rheumatic and neoplastic diseases. Arthritis Rheum. 22: 36-42, 1979.
18. Steel DM, Whitehead AS, The major acute phase reactants : C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol. Today. 15 : 81-8, 1994.
19. De Beer FC, Fagan EA, Hughes GRV, Mallya R, Lanham JG, Pepys MB, Serum amyloid-A protein concentration in inflammatory diseases and its relationship to the incidence of reactive systemic amyloidosis, Lancet ; 31 : 231-4,1982.
20. Maury C.P., Teppo A.M. & Wegelius O. Relationship between urinary sialylated saccharides, serum amyloid A protein, and C-reactive protein in rheumatoid arthritis and systemic lupus erythematosus, Ann. Rheum. Dis. 41 (3): 268-71,1982.
21. Ganapathi MK, May LT, Schultz D, Role of interleukin-6 in regulating synthesis of C-reactive protein and serum amyloid A in human hepatoma cell lines, Biochem. Biophys. Res. Common. 157 : 271-7, 1988.
22. Grehan S, Uhlar CM, Sim RB, Herbert J, Whitehead AS : Expression of biologically active recombinant mouse IL-1 receptor antagonist and its use in vivo to modulate aspects of the acute phase response, J Immunol. 159 : 369-78, 1997.
23. Rokita H, Loose LD, Bartle LM, Sipe JD, Synergism of interleukin 1 and interleukin 6 induces serum amyloid A production while depressing fibrinogen: a quantitative analysis, J Rheumatol. 21: 400-5, 1994.
24. Kushner I, The acute phase response : An overview, Methods Enzymol. 163 : 373-83, 1988.
25. Lange U, Boss B, Teichmann J, Klor HU, Neeck G: Serum amyloid A an indicator of inflammation in ankylosing spondylitis, Rheumatol Int. 19(4):119-22, 2000.
26. Stadnyk A, Gauldic J : The acute phase protein response during parasitic infection, Immunol Today. 12 : A7-12, 1991.
27. Husby G, Marhaung G, Dowton B, Sletten K, Sipe JD, Serum amyloid A (SAA): Biochemistry, genetics and pathogenesis of AA amyloidosis, Amyloid. 1: 119-137, 1994.
28. Demin AA, Semenova LA, Sentiakova TN, Poliachenko EA, Smirnov VV, Potaliukova EV, Demina LM, Amyloidosis in systemic lupus erythematosus, Klin Med (Mosk). Feb;70(2):61-6, 1992.
29. Maury CP, Teppo AM, Antibodies to amyloid A protein in rheumatic diseases, Rheumatol Int. 8(3):107-11, 1988.
30. Yamada T. Genetic effects on serum concentrations of serum amyloid A protein. Clin Chem. 50(5):978-9, 2004.
31. Okuda Y, Yamada T, Takasugi K, Takeda M, Nanba S, Onishi M, Miyamoto T, Inoue Y, Serum amyloid A (SAA) 1, SAA 2 and apolipoprotein E isotype frequencies in rheumatoid arthritis patients with AA amyloidosis, Ryumachi. 39(1):3-10, 1999.
32. Yamada T, Okuda Y, Takasugi K, Itoh K, Igari J, Relative serum amyloid A (SAA) values: the influence of SAA1 genotypes and corticosteroid therapy in Japanese patients with rheumatoid arthritis, Ann Rheum Dis. 60:124-127, 2001.
33. Kumon Y, Suehiro T, Hashimoto K, Sipe JD. Dexamethasone, but not IL-1 alone, upregulates acute-phase serum amyloid A gene expression and production by cultured human aortic smooth muscle cells. Scand J Immunol, 53(1):7-12, 2001.
34. Steel DM, Whitehead AS, The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein, Immunol Today. 15(2):81-8, 1994.
35. Baumann H, Gauldie J. The acute phase response, Immunol Today. 15(2):74-80, 1994.
36. Baumann H, Gauldie J. The acute phase response, Immunol Today. 15(2):74-80, 1994.
37. Manzi S, Seizer F, Sutton-Tyrrel K, Fitzgerald SG, Raire JE, Tracy RP, Kuller LH, Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus, Arthritis Rheum. 42 (1) : 51-60, 1999.
38. Kaysen G.A, The microinflammatory state in uremia: causes and potential consequences. J. Am. Nephrol. 12(7):1549-57, 2001.
© 2004 Egyptian Dermatology Online Journal
|
