Abstract:
Degos disease (malignant atrophic papulosis) is a rare disease with an unknown etiology. It affects skin as well as other organs. Cutaneous lesions are in the form of red papules that develop porcelain white centers and telangiectatic borders. Abnormal coagulation caused by disturbed endothelial function may be the immediate causative factor. No effective treatment has yet been found. We are reporting a case of Degos disease including the diagnostic procedures as well as the prognostic criteria. Clinical Synopsis
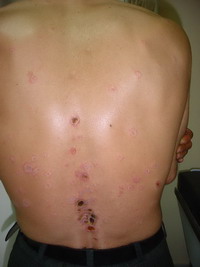
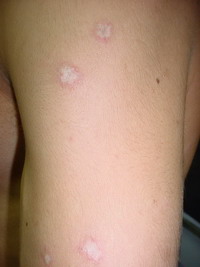
A 38-year- old male presented by a remittent eruption of small papules (3 or 4 each week) without complete disappearance of older one . The papules were erythematous, less than one centimeter in diameter with an irregular distribution on the trunk & proximal extremities, sparing the face, palms & soles. Older lesions were seen with atrophic porcelain-white centers that surrounded by telangiectasia
{Fig1}. Two months later the patient complained of abdominal distention & disturbed bowel movement and gave a history of treatment by topical & systemic corticosteroid with no response.
| |
| Fig 1: Lesions
on the back and left arm |
|
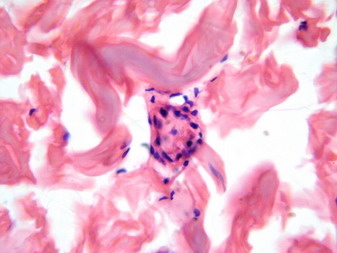
Pathological Examination: Two skin biopsies were taken; one from the active lesion and the other from a healed one. The first biopsy revealed epidermal ulceration
{Fig 2a} & wedge shaped area of dermal collagen necrosis {Fig 2b} with its base towards the epidermis &apex in lower dermis.
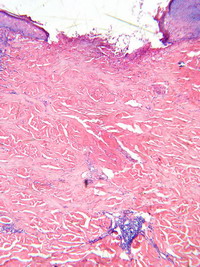 | Fig
2a: Epidermal ulceration |
|
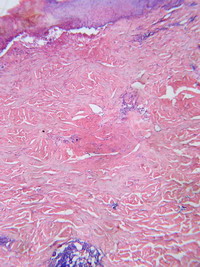 Fig
2b: wedge- shaped
area of dermal collagen necrosis. |
|
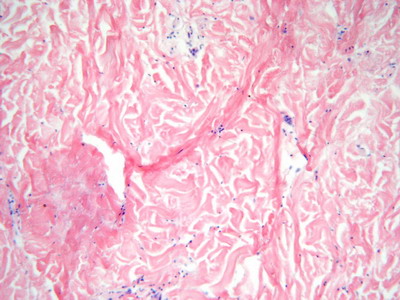
Step sectioning revealed thrombosed blood vessel {Fig 3} in the reticulate dermis deep to the area of the wedge-shaped infarction.
| |
| Fig 3:
Obliterated blood vessels. |
|
The second biopsy showed epidermal atrophy, fibrosis and mucin deposition {Fig 4}.
Laboratory investigations: Blood samples were examined and revealed platelet aggregation to the three aggregating agents and showed normal lag phase:
- Platelet aggregation to ristocetin was 83.7% (60-120).
- Platelet aggregation to collagen was 80.4% (60-120).
- Platelet aggregation to ADP was 85.3% (60-120).
- Platelet adhesion was 89 % (75-95).
Stool examination revealed the presence of occult blood. GIT endoscopy and GIT biopsy: A GIT endoscopy was done and a picture suggestive of vasculitis was reported. A GIT biopsy was then performed and a perivascular lymphocytic infiltration was revealed.
Other investigations: CT scan of the brain revealed no abnormalities and immune-histochemical tests revealed -ve IgG.
Discussion
Degos disease was first described by Robert Degos in 1942 and it may occur in two forms; a limited benign form, and multi-system lethal form. It is characterized by infarctive lesions that can occur in skin or other organ leading to necrosis and bleeding. No age is immune (although more common in adulthood). In 50% of cases GIT is affected in the form of nausea, vomiting, bowel movement & bleeding while in 20 % of cases C.N.S. is affected in the form of headache, dizziness and paraplegia. Ocular affection including diplopia, ptosis and posterior sub-capsular cataract were also reported. Other systems like the heart, lungs, liver and kidneys can be affected. Systemic affection occurs few weeks to years after cutaneous affection but may precede the skin lesion on rare cases. Our patient has the characteristic cutaneous lesions that were proved by biopsy to be small infarcts only few weeks before GIT symptoms started to manifest. A familial variant of Degos disease has been described by Katz et al., 1997 so the patient, as well as his family members, should be closely followed up and signs of external or internal bleeding should be closely watched for early intervention to minimize complications. References
1. Degos R, Malignant atrophic papulosis, Br J Dermatology. 100: 21- 35, 1979.
2. Ball E, Newburger A, Ackerman AB, Degos' Disease: A Distinctive Pattern of Disease, Chiefly of Lupus Erythematosus, And Not a Specific Disease per se, Am J Dermatopathol. 25 (4): 302-320, 2003.
3. Katz SK, Mudd LJ, Roenigk HH, Malignant atrophic papulosis (Degos disease) involving three generations of a family, J Am Acad Dermatol. 37: 480- 484, 1997.
4. Al-Smadi RM, Abu-Jamous F , Omeish I, Degos disease in a 24-year-old Jordanian male, Eastern Mediterranean Health Journal. 6 (1): 194- 196, 2000.
5. Scheinfeld NS, Degos Disease, eMedicine, February 3, 2005 http://www.emedicine.com/derm/TOPIC931.HTM© 2008 Egyptian Dermatology Online Journal |