|
|
Abstract:
We present three cases of Hyper IgE syndrome who presented with recurrent episodes of lower respiratory tract infection, skin lesions and facial dysmorphism. Evaluation revealed high levels of serum IgE. The screening of parents was done for the same, which was normal. The patients were managed symptomatically and are under follow up. This case series highlights the unusual and varied presentation of Hyper IgE syndrome which a clinician must have in mind while suspecting any case of immunodeficiency. Introduction:
The hyper-IgE syndrome is a rare immunodeficiency syndrome characterized by recurrent skin and pulmonary abscesses and extremely elevated levels of IgE in the serum [1]. We are reporting our experience with three cases of hyper- IgE syndrome that we encountered at our institution.
Case Report 1: A 4 and half years old male child was brought with history of recurrent episodes of cough and wheezing since early infancy. The patient was also diagnosed to have atopic dermatitis at 8 months of age. There was history of hospitalization for bronchopneumonia at one year of age. At three years of age he had received a 6 months course of antituberculous treatment (ATT) for suspected primary complex. A Chest X ray & CT at 4 yrs of age revealed left lung cyst for which a left lobectomy was done. On examination, he was noted to have a broad forehead with widely spaced eyes, coarse facial features (Fig1) and clubbing. The child had multiple subcutaneous abscesses and scars of old healed furuncles over the skin.
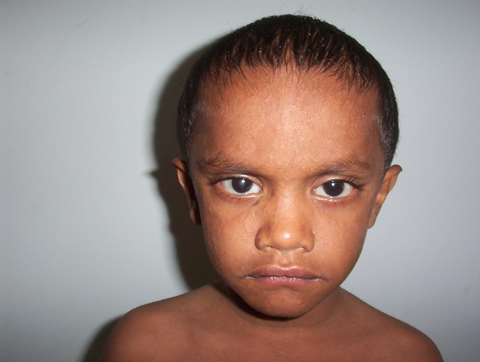 | Fig 1:
Case 1 showing the broad forehead & nose with widely spaced eyes. |
|
Routine hemogram, serum biochemistry, liver function and renal function tests were within normal limits. A chest X-ray showed patchy consolidation in both upper zones with pleural thickening. A contrast enhanced computed tomography (CECT) chest showed mediastinal lymphadenopathy with bilateral pulmonary infiltrates and cystic lesions in the left lung (Fig 2). Sputum for acid-fast bacilli (AFB) and Mantoux tests were negative. Sputum culture showed a growth of Staphylococcus aureus. Based on the culture and sensitivity pattern, he was managed with the appropriate antibiotics. In view of his present and past clinical profile of recurrent skin and pulmonary infections with dysmorphic facial features, he was further investigated to rule out an immune-deficiency disorder. An Immunoglobulin profile revealed IgG of 1486mg/dl (Ref range 550-1400), IgM of 287 mg/dl (Ref range 40-95), IgA of 190 mg/dl (Ref range 60-200) and IgE of 2414 IU/ml (Reference range 0-50). Serum IgE levels of the child's parents were tested wherein the father IgE was 310 IU/ml (Ref range 0-180). The IgE levels in the mother were within normal limits.
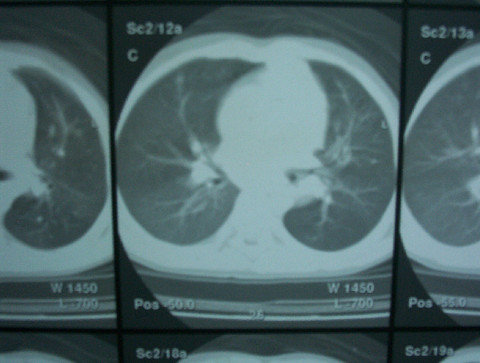 | Fig
2:
Chest CT scan showing bilateral mediastinal lymphadenopathy. |
|
Cases 2 and 3: A 14 years old girl was brought with the complaints of recurrent purulent discharge from both ears for the past 7 years, multiple hyperpigmented skin lesions over face, forearms and both legs for 2 years and recurrent erosions over lower lip for 2 years. Family history revealed that her elder brother aged 19 years had similar recurrent skin lesions and had been managed for empyema and psoas abscess earlier. He had also undergone a lobectomy for lung cyst at the age of 9 years. On examination, her height and weight were in the 3rd percentile. She had coarse facies with broad forehead. Dermatological examination revealed multiple hyperkeratotic, hyperpigmented papules and pustules distributed over both arms and legs with skip areas of normal skin in between. Oral cavity showed diffuse erosions and depigmentation over lips and poor enamelization and crowding of teeth. Ear examination revealed bilateral chronic suppurative otitis media with conductive deafness. An immunoglobulin profile revealed markedly elevated IgE levels at 2138 IU/ml (ref range-<100 IU/ml). Other immunoglobulin levels were within the normal range. Testing of the immunoglobulin profile of the patient's brother was also done which revealed marked elevation of IgE levels (2785 IU/ml). Discussion:
Hyper IgE syndrome (HIES) with recurrent infections (Job's syndrome) is a rare idiopathic primary immunodeficiency disease characterized by the triad of elevated serum IgE levels (>2000 IU/ml), recurrent cutaneous abscesses and recurrent sinopulmonary infections [2]. The etiology & pathophysiology of Job syndrome are not completely understood. The mechanisms responsible for increased IgE production are unknown. There is evidence to suggest that these patients have T cell defects, which result in defective antibody responses to specific antigens [3]. HIES is a multisystem phagocytic disorder that affects the dentition, skeleton, connective tissue and immune system. It is inherited as a single locus autosomal dominant trait with variable expressivity. The disease focus for HIES has been mapped to the proximal region of chromosome 4q [4]. Usually IgE levels in HIES exceed 2000 IU/ml. This was also seen in our cases. Recently, an autosomal recessive variant has been described in which the IgE level is lower than reported earlier [5]. Dermatitis is present in more than 80% of the patients of HIES and usually begins at 2 months to 2 years of age. The skin and soft tissue infections present as cellulitis, furunculosis, paronychia, suppurative adenitis and deep soft tissue 'cold' abscesses. Severe pulmonary infections caused by either S. aureus or H. influenzae are common. Empyema may complicate pneumonia and there is a high propensity for bronchiectasis and pneumatoceles. Otitis externa, chronic otitis media, chronic mastoiditis, dental and periodontal diseases are common. Mucocutaneous candidiasis and tinea unguium are also observed in HIES [6]. Some non- immunological features of HIES have been reviewed by Grimbacher et al [1] which include failure or delay of shedding of primary teeth, recurrent bone fractures, joint hyper-extensibility, scoliosis and distinctive facial appearance. No definitive therapy is available for the treatment of Hyper IgE syndrome. Therapy is mainly directed at prevention and management of infections by using sustained systemic antibiotics and antifungals along with topical therapy for eczema and drainage of abscesses. Anti-staphylococcal antibiotic prophylaxis is useful. Interferons, immunoglobulin supplementation, or low-dose cyclosporine A have been reported to benefit selected patients, but they are not generally indicated [7]. Many patients who are undergoing regular monitoring and receiving appropriate treatment will live beyond the age of 50 yrs. Death is often due to infectious complications References
1. Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O'Connell AC, Puck JM. Hyper-IgE syndrome with recurrent infection: an autosomal dominant multisystem disorder. N Engl J Med. 1999; 340(9): 692- 702
2. Muhammed K. Hyper IgE syndrome: report of two cases with moderate elevation of IgE. Indian J Dermatol Venerol Leprol. 2005; 71(2): 112- 114.
3. Donata Vercelli, Haifa H. Jabara, Charlotte Cu Rundles, John S. Abrams, David B. Lewis, Jeff Meyer, L Schneider, Donald Y. M. Leung, Raif S. Geha. Regulation of Immunoglobulin (1g) E Synthesis in the Hyper-IgE Syndrome. J Clin Invest.1990; 85(5): 1666- 1671.
4. Segal BH, Holland SM. Primary phagocytic disorder of childhood. Pediatr Clin North Am 2000; 47: 1311- 1338.
5. Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: A distinct disease entity. J Pediatr 2004; 144: 93- 99.
6. Paller AS. Cutaneous manifestations of Non-AIDS immunodeficiency. In: Moschella SL, Hurly HJ, editors. Dermatology. Philadelphia: WB Saunders Company; 1992. p. 356- 360.
7. Mary Wakim, Micheal Alazard, Ava Yajima, David Speights, et al. High dose intravenous immunoglobulin in atopic dermatitis and hyper-IgE syndrome. Ann Allergy Asthma Immunol 1998; 81: 153- 158.© 2010 Egyptian Dermatology Online Journal |
