|
|
Abstract
The drug rash with hyper-eosinophilia and systemic symptoms (DRESS) syndrome is a severe drug-induced hypersensitivity syndrome. Pyrazinamide, an antitubercular drug, has been described as an uncommon cause of DRESS syndrome. We report a 45- year-old woman suffering from DRESS syndrome 10 days after pyrazinamide exposure. She had a skin eruption, weakness, fever, and lymph node enlargement. Laboratory analysis revealed leukocytosis, lymphocytosis, hyper-eosinophilia, thrombocytopenia, inflammation, hepatic cytolysis, and cholestasis. Urine examination showed protein ++, and aseptic pyuria indicating most likely an interstitial nephritis. She fully recovered after drug withdrawal. To our knowledge, this represents the first case report of DRESS syndrome secondary to pyrazinamide since its commercialization. Introduction
Tuberculosis, whatever its localization, is an infectious disease which can be totally cured by combining antitubercular drugs. Current therapeutic regimens with isoniazid, rifampin, pyrazinamide, ethambutol, and streptomycin have proved successful in treating tuberculosis. However, they are associated with a high rate of adverse effects that can lead to therapeutic failure. Understanding the nature and the severity of these adverse effects allows for their appropriate management. The major adverse effect related to pyrazinamide is liver injury which is rare but sometimes lethal. Pyrazinamide may induce numerous cutaneous adverse drug reactions as well as diffuse pruriginous maculopapular rash and it has been described as an uncommon cause of DRESS syndrome. DRESS syndrome is a life-threatening pattern of a drug hypersensitivity reaction. The syndrome's complete form combines a severe febrile skin eruption (usually the first sign), hyper-eosinophilia, atypical lymphocytes, and organ involvement, such as hepatitis, myocarditis, interstitial nephritis, and interstitial pneumonitis. Lymphadenopathy and hepato-splenomegaly may be present, as may arthritis and synovitis [1,2,3].
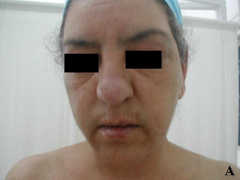
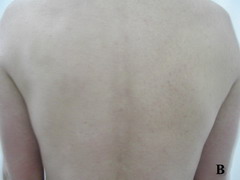
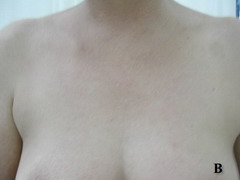
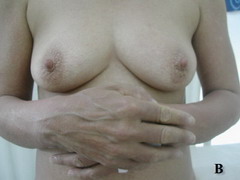
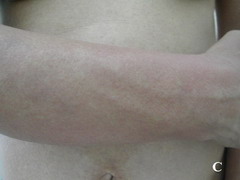
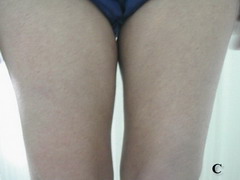
Case report A 45- year-old woman was admitted for asthenia, anorexia, weight loss, night sweats and fever of 38.5° C. The symptoms had been present for 3 months. The physical examination at admission found sub-centimetre lymph nodes of the jugular-carotid chain which were firm, mobile and painless. The intra-dermal tuberculin test produced a 18-mm phlyctenular induration. The liver and spleen were not enlarged. Laboratory tests showed inflammation (erythrocyte sedimentation rate, 50mm/h and C-reactive protein, 18mg / l). Sputum and urine tests were negative for the tubercle bacillus. The CT scan showed mediastinal lymphadenopathy associated with pericardial effusion and intra-abdominal and bilateral pulmonary nodules. The lymph node biopsy was performed and histology showed an epithelioid and giant-cell granuloma with caseation. The diagnosis was made as disseminated tuberculosis and treatment consisted of four antitubercular drugs (rifampin, isoniazid, pyrazinamide, and streptomycin). The patient developed on the tenth day of her treatment, a pruriginous maculopapular erythema initially involving the face (Fig 1 A), trunk (Fig 1B), and limbs (Fig 1C) associated with a characteristic swelling of the lips (Fig 1A); a fever of 40° C; a major weakness; and enlargement of the liver, spleen, and several peripheral lymph nodes.
| |
| |
| |
| Fig 1:
Maculopapular erythema initially involving the face with a
characteristic swelling of the lips (A), trunk (B), and limbs (C). |
|
Laboratory analysis revealed leukocytosis (14,500/ mm ³), lymphocytosis (5000/ mm ³), hyper-eosinophilia (2300 / mm ³), thrombocytopenia (70 000/mm ³), and inflammation (erythrocyte sedimentation rate, 72mm/h and C-reactive protein, 35mg / l). Other laboratory findings were as follows: moderate hepatic cytolysis (ALAT, 141 IU/l [N,lt;40IU/l]; and ASAT, 111 IU/l [N,lt;40IU/l]), and cholestasis (gamma GT, 572 IU/l [N,10-40IU/l]; and alkaline phosphatase, 987 IU/l [N,60-275IU/l]). Urine examination showed protein ++, and aseptic pyuria indicating most likely an interstitial nephritis. No bacteria were found in blood, or urine. No evidence of lymphoma or other haematological malignancies was detected by laboratory tests or histological studies. Bone marrow aspiration was normal. Findings were negative for serological tests for hepatitis B and C viruses, and human immunodeficiency virus. The diagnosis of hypersensitivity to antitubercular drugs was made. Antitubercular drugs were withdrawn and then reintroduced one by one (at least to the most incriminated). The re-introduction of pyrazinamide, three days after that of isoniazid, was marked by the reappearance of symptoms, this molecule was then objected to the suggestive trend marked by the disappearance of symptoms after drug withdrawal, recurrence of symptoms immediately after the resumption of pyrazinamide and pharmaco-vigilance survey which confirmed the accountability of pyrazinamide. The evolution was marked by the disappearance of skin lesions and lymph nodes, the normalisation of blood counts and liver function tests, three weeks after antitubercular drugs withdrawal. Anti-tuberculosis treatment, excluding pyrazinamide, was started after normalization of liver function. The clinical outcome was favourable. Discussion
This patient presented with a severe febrile skin eruption associated with systemic involvement including lymph nodes, leukocytosis, lymphocytosis, hyper-eosinophilia, thrombocytopenia, inflammation, hepatic cytolysis, and cholestasis following drug intake. We believe that one of the antitubercular drugs was responsible for the DRESS syndrome. The diagnosis was suggested by the disappearance of symptoms after pyrazinamide withdrawal, recurrence of symptoms immediately after the resumption of pyrazinamide which was confirmed by pharmaco-vigilance survey. The most frequently incriminated drugs are the aromatic anticonvulsants and sulfonamides. The syndrome has been also associated with the use of captopril and calcium channel blockers, allopurinol, ranitidine, thalidomide, ibuprofen, gold salts, minocycline, sulfasalasine, and nevirapine [1,4]. The diagnosis of DRESS syndrome requires the simultaneous presence of three criteria [1]: (a) drug-induced skin eruption, (b)
eosinophilia ≥ 1.5 x 109/l or atypical lymphocytes, and (c) at least one of the following systemic abnormalities: enlarged lymph nodes at least 2 cm in diameter, hepatitis
(transaminases ≥ 2N), interstitial nephropathy, interstitial lung disease, or myocardial involvement. The syndrome develops 2-6 weeks after treatment initiation [4]. The skin disease is characterized by an infiltrated maculopapular eruption and facial edema, often more marked in the peri-orbital regions. The lesions develop first on the trunk then spread to the rest of the body. The initial edematous maculopapular lesions convert to blisters, vesicles or pustules [5]. The histological finding lack specificity, consisting mainly of a lymphocytic inflammatory infiltrate and, less prominently, of eosinophils in the papillary dermis [5]. DRESS syndrome continues to carry a high mortality rate of about 10% [1,6]. Liver involvement with necrosis leading to hepatic failure is the principle cause of mortality [4]. Pericarditis, myocarditis, and meningoencephalitis are less common [4]. The multi-visceral involvement also differentiates the DRESS syndrome from cutaneous drug eruptions. The patho-physiology of DRESS syndrome has not been fully elucidated. The prevailing hypothesis holds that the causative drug induces hypersensitivity as a result of abnormalities in the production and detoxification of its active metabolites. These abnormalities may be related to a genetic predisposition, as the risk is increased in patients with a positive family history for DRESS syndrome, in slow acetylators, and in blacks [7,8,9,10,11]. HHV6 has been incriminated recently as a cofactor in the development of DRESS syndrome [12,13]. This cofactor effect may occur either during primary HHV6 infection or during reactivation of HHV6 infection from particles that remain dormant in lymphocytes and monocytes. The treatment is not standardized at present. The suspected drug should be stopped immediately. Delaying this measure may be associated with a worse prognosis [14]. Several weeks are needed to determine whether a recovery has been achieved. Glucorticoids remain the most widely used agents, although dosages vary widely across case reports. Glucorticoid therapy is mandatory in severe cases and can produce a dramatic improvement. Dependency on glucocorticoids may develop, however, with a risk of recurrent clinical or laboratory test abnormalities during the tapering process. Treatment with high-dose intravenous N-acetylcysteine (NAC) has been proposed. NAC is a known precursor of glutathione, a molecule involved in the detoxification of several drugs [15,16]. Conclusion
Cases of DRESS syndrome in patients undergoing treatment with pyrazinamide should be reported to pharmaco-vigilance centers to ensure that the frequency of this adverse reaction is not underestimated. It is important to recognize this entity because it is a potentially life-threatening condition. Prompt withdrawal of incriminating drugs is mandatory. References
1. Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (Drug Rash With Eosinophilia and Systemic Symptoms: DRESS). Semin Cut Med Surg 1996; 1: 250- 257.
2. Bonnetblanc JM.Drug hypersensitivity syndrome. Dermatology 1993; 187: 84- 85.
3. Yamakado S, Yoshida Y, Yamada T, Kishida T, Kobayashi M, Nomura T. Pulmonary infiltration and eosinophilia associated with sulfasalazine therapy for ulcerative colitis: a case report and review of literature. Intern Med 1992; 31: 108- 113.
4. Ichiche M, Kiesch N, De Bels D. DRESS syndrome associated with HHV-6 reactivation. Eur J Intern Med 2003; 14: 498- 500.
5. Michel F, Navellou JC, Ferraud D, Toussirot E, Wendling D. DRESS syndrome in a patient on sulfasalazine for rheumatoid arthritis. Joint Bone Spine 2005; 72: 82- 85.
6. Bouyssou-Gauthier ML, Bedane C, Boulinguez S, Bonnetblanc JM. Photosensitivity with sulfasalazopyridine hypersensitivity syndrome. Dermatology 1999 ; 198 : 388- 390.
7. Pinals RS. Sulfasalazine in the rheumatic diseases. Semin Arthritis Rheum 1988; 17: 246-259.
8. Mc Kenna KE, Burrows D. Leucopenia , thrombocytopenia and lymphoadenopathy associated with sulphasalazine. Clin Exp Dermatol 1994; 19 : 419- 420.
9. Delboy H. Les effets indésirables de la salazosulfapyridine. Sem Hop Paris 1992; 68:180- 187.
10. Rieder MJ, Shear NH, Kanee A. Prominence of slow acetylator phenotype among patients with sulphonamide hypersensitivity reactions. Clin Pharmacol Ther 1991; 49: 13- 17.
11. Nielsen OH. Sulfasalazine intolerance: a retrospective survey of the reasons for discontinuing treatment with sulfasalazine in patients with chronic inflammatory bowel disease. Scand J Gastroenterol 1982; 17: 389- 393.
12. Tohyama M, Yahata Y, Yasukawa M, Inagi R, Urano Y, Yamanishi K, et al. Severe hypersensitivity syndrome due to sulfasalazine associated with reactivation of human herpesvirus 6. Arch Dermatol 1998; 134: 1113- 1117.
13. Suzuki Y, Inagi R, Aono T, Yamanishi K, Shiohara T. Human herpes virus 6 infection as a risk factor for the development of severe drug-induced hypersensitivity syndrome. Arch Dermatol 1998; 134: 1108- 1112.
14. Queyrel V, Catteau B, Michon- Pasturel U, Fauchais AL, Delcey V, Launay D, et al. DRESS syndrome à la sulfasalazine et à la carbamazépine : à propos de deux cas. Rev Med Intern 2001 ; 22: 582- 586.
15. Gabay C, De Bandt M, Palazzo E. Sulphasalazine-related life-threatening side effects: is N-acetylcysteine of therapeutic value? Clin Exp Rheumatol 1993; 11: 417- 420.
16. Hansen RM, Varma RR, Hanson GA. Gold induced hepatitisand pure red cell aplasia. Complete recovery after corticosteroid and N-acetylcysteine therapy. J Rheumatol 1991; 18: 1251- 1253.© 2010 Egyptian Dermatology Online Journal |