|
|
Abstract
Pachyonychia congenita (PC) is a rare, but well characterized autosomal
dominant disorder of keratinization in which different mutations affect
the genes of keratins K6a, K6b, K16 and K17. A three year old male child
presented with over curvature of nails and spiny papules over the body since
early infancy. Further examination revealed small area of leukokeratosis
in the mouth and palmoplantar keratoderma. There was no hair and laryngeal
involvement which can be due to incomplete penetrance of genes. The case
is being reported for its rare occurrence.
Introduction
Pachyonychia congenita (PC) is a group of inherited ectodermal dysplasias
in which the most prominent feature is hypertrophic nail dystrophy [1].
Muller and Wilson described the first case of pachyonychia congenita (PC)
and one year later similar case was reported by Jadassohn and Lewandowsky
[2].There are mainly two types of pachyonychia
congenita (PC): PC1 (Jadassohn Lewandowsky syndrome) and PC2 (Jackson Lawler
syndrome). Here we report a case of three-year-old male child with features
suggestive of Jadassohn Lewandowsky syndrome.
Case report:
A three-year-old male child born to non-consanguineous parents
presented with papular, scaly lesions over the trunk with thickening of
all nails and soles at pressure points since early infancy. Patient had
burning sensation in the tongue and mild hoarseness of voice, for the last
six months. History of mild hyperhidrosis was present and developmental
milestones were normal. There was no history of similar complaint in the
family. Systemic examination revealed no abnormality. On cutaneous examination:
generalized, follicular, erythematous, papular lesions were present mainly
over the upper trunk, elbows, knees, buttocks, and anterior ankles (Fig.
1). Thick, yellowish, hyperkeratotic plaques were present over the pressure
points of the soles (Fig. 2). Palms also showed thick, hyperkeratotic papules.
On nail examination: all the twenty nails were wedge shaped, yellow, and
thickened with subungual hyperkeratosis (Fig.3). In the oral cavity: angular
cheilitis and whitish plaque was present over the dorsum of the tongue extending
on to the lateral margins (Fig.4). Otolaryngeal, ophthalmic and hair examination
was normal.
All routine investigations were within normal limits. KOH examination
of nails was negative for fungus. Biopsy of the lesions could not be done
due to unwillingness of the parents. With all these clinical findings, diagnosis
of PC1 (Jadassohn Lewandowsky syndrome) was made and patient was put on
oral vitamin A, keratolytic agents and emollients.
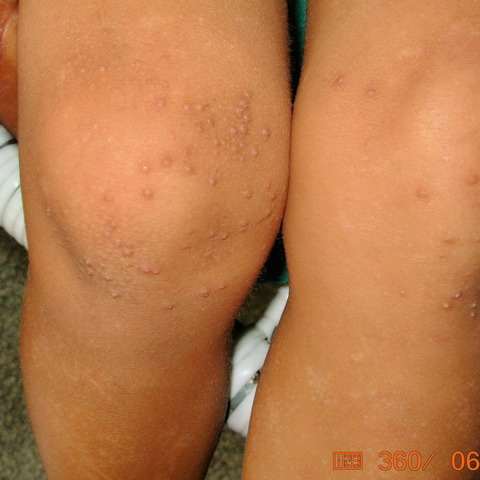 | Fig 1:
Showing follicular keratosis on the knees. |
|
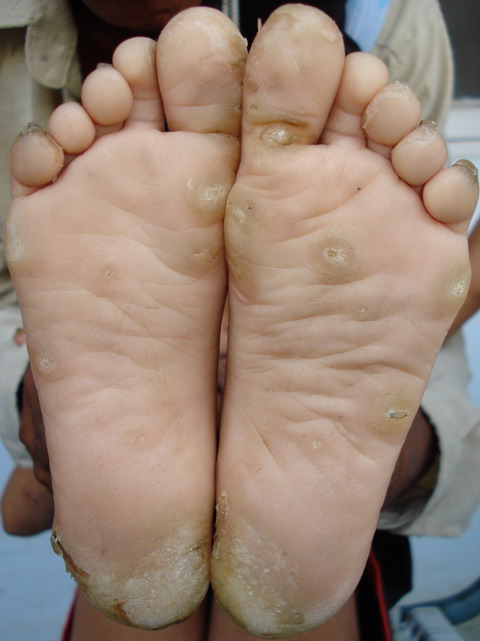 | Fig
2: Showing focal keratoderma on soles. |
|
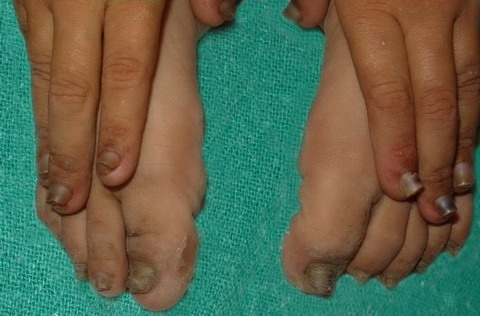 | Fig
3: Showing pachyonychia in hands and feet. |
|
|

|
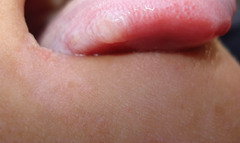
|
| Fig 4: Showing leukoplakia on tongue and angular cheilitis of
mouth. |
|
Discussion
Pachyonychia congenita (PC) is a rare, autosomal dominant disorder characterized
by triad of subungual hyperkeratosis with accumulation of hard keratinous
material beneath the distal portion of the nails, lifting the nails from
the nail bed, keratosis palmaris et plantaris with thick callosities, especially
on the soles and thick white areas on the oral mucosa[3].
Other associated features which may occur include keratosis pilaris, hyperkeratotic
follicular papules on the sites of friction, hair abnormalities and hyperhidrosis
of the palms and soles. These disorders have been suggested to be due to
mutations in paired keratins, K6a/K16 (in PC1) and K6b/K17 (in PC2) [4,5].
According to these mutations, various clinical variants have been described
[6]:
Type 1- PC1: Jadassohn Lewandowsky syndrome which is characterized by
focal palmoplantar keratoderma (PPK), follicular keratosis mainly on the
trunk and oral leucokeratosis.
Type 2-PC2: Murray Jackson Lawler syndrome is the most common form associated
with mild focal PPK, pili torti, natal teeth and multiple epidermal cysts.
Angular cheilitis, hoarseness, follicular keratosis may be present in both
types,
Type 3: includes combined features of types 1 and 2 with angular chielitis,
corneal dyskeratosis, and cataracts.
Type 4: includes features of type 1 and type 3 with laryngeal lesions,
hoarseness of voice with mental retardation, hair abnormalities and alopecia.
Other rare variants include pachyonychia congenita with only nail involvement
and pachyonychia tarda that is pachyonychia congenita with onset in teenage
years [7,8].Complications
like respiratory distress due to laryngeal leucokeratosis and acro osteolysis,
malignant changes in palmoplantar lesions can occur in pachyonychia congenital
[6].
Treatment options for PC fall into four broad categories:
(1) Non- invasive (mechanical) e.g. abrasion with some hand tool
(2) Invasive (surgical) e.g. electrofulgration, excision
(3) Chemical methods using urea, propylene glycol, alpha hydroxy acid
(4) Pharmacological (vitamin A, retinoids), all basically targeted at
reducing the hyperkeratosis involving different sites [9].
When the familial mutation is known, genetic counseling can be done and
if required, prenatal diagnosis can be done at early stage of pregnancy
by chorionic villi biopsy [10].
For all these reasons, a patient with pachyonychia congenita should be
thoroughly investigated and treated accordingly.
References
1. Irvine AD, McLean WH. Human keratin diseases: the increasing
spectrum of disease and subtlety of the phenotype-genotype correlation,
Br J Dermatol1999; 140: 815- 828.
2. Feinstein A, Friedman J, Schewach-Millet M, Pachyonychia
congenita, J Am Acad Dermatol 1998; 19: 705- 711.
3. Johnson BL Jr, Yan AC. Congenital diseases (Genodermatosis).
In: Elder DE (eds). Lever's Histopathology of the Skin, 10th edition. Philadelphia,
Lippincott Williams & Wilkins, 2009: P138.
4. McLean WH, Rugg EL, Lunny DP et al., Keratin 16 and keratin
17mutations cause pachyonychia congenita, Nat Genet 1995; 9: 273- 278.
5. Leachman SA, Kaspar RL, Fleckman P et al., Clinical and
pathological features of pachyonychia congenita, J Invest Dermatol Symp
Proc. 2005; 10: 3- 17.
6. Murugesh SB, Reddy S, Ragunatha S et al., Acro-osteolysis:
a complication of Jadassohn-Lewandowsky syndrome. Int J Dermatol 2007; 46:
202- 205.
7. Bansal A, Sethuraman G, Sharma VK. Pachyonychia congenita
with only nail involvement, J Dermatol 2006; 33(6): 437- 438.
8. Paller AS, Moore JA, Scher R. Pachyonychia congenital
tarda: a late onset form of pachyonychia congenita, Arch Dermatol 1991;
127: 701- 703.
9. Milstone LM, Fleckman P, Leachman SA et al.,Treatment
of pachyonychia congenita, J Investig Dermatol Symp Proc. 2005; 10(1): 18-
20.
10. Smith FJ, McKusick VA, Nielsen K et al., Cloning of
multiple keratin 16 genes facilitates prenatal diagnosis of pachyonychia
congenita type 1, Prenat Diagn. 1999; 19(10): 941- 946.
© 2010 Egyptian Dermatology Online Journal
|
