|
|
Abstract
Prolidase deficiency is a rare inborn disorder of collagen metabolism characterized by chronic recurrent skin ulceration. A twenty five- year-old male with clinical features and laboratory criteria fulfilling the diagnosis of prolidase deficiency is presented in view of rarity of the condition
Introduction
Prolidase deficiency is a rare disorder inherited through an autosomal recessive gene. The hallmark of this disorder is the presence of characteristic face with saddle nose, mental retardation and chronic recurrent cutaneous ulcers that are recalcitrant in healing. The ulcers result from impaired recycling of proline which constitute 20% of amino acid in collagen and is important for tissue repair. Other features that are seen include frequent infections, partial deafness, visual disturbances and joint dislocation[1] . Case report
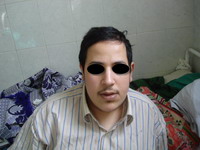
A 25 year- old Egyptian male presented to the outpatient Clinic of Dermatology and Venereology Department of Tanta University Hospitals in October, 2008. He complained of ulcer on the right leg since 3 years and another one on the dorsum of left foot since 2.5 years . The ulcers were slightly painful with history of delayed healing. There are oozing and crusted skin lesions on both palms and forearms with pruritus 7 months ago. He was suffering from difficulties in learning and unsocial behavior. He was hypertensive. No history of other systemic diseases or recent drug intake. On general examination, he appeared overweight, short stature with leaning forward deformity in his back. He had an abnormal low hair line (Fig1a)
|
|

|
| Fig 1a: A 25 years male patient. He had an
abnormal low hair line. He appeared over weight, short
stature with leaning forward deformity in his back. |
|
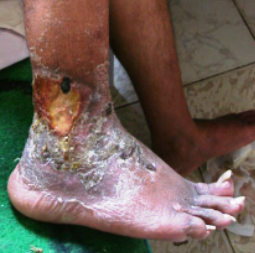
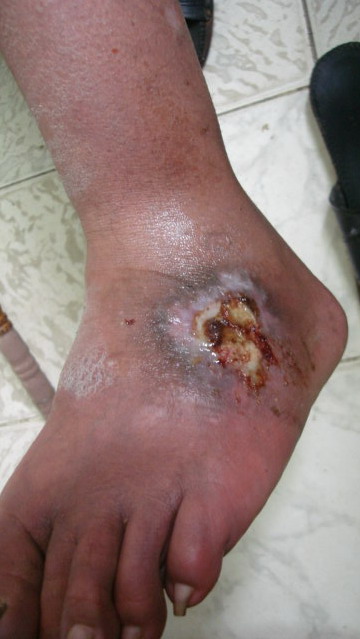
On local examination, deep well defined irregular ulcer 4x6 cm in diameter just above lateral malleolus of the right leg, It is characterized by irregular borders with prominent granulation tissue and purulent exudates and surrounded by dermatitic changes with crustation and hyperpigmentation. The other ulcer on the dorsum of the left foot was smaller and more superficial showing signs of healing
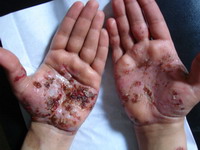
(Fig 1b).. There are eczematous plaques on both palms and forearms (Fig 1c) and lymphedema in the right leg. No lymph node enlargement. Abdominal examination revealed nothing abnormal.
|
|
|
|
| Fig 1b: Deep well defined irregular ulcer above
lateral malleolus in the right leg and left foot, surrounded
by dermatitic changes with crustation and hyperpigmentation. |
|
|
|
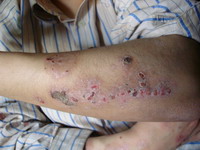
|
| Fig 1c: Oozing and crusted eczematous plaques on
both palms and forearms. |
|
Laboratory investigations revealed microcytic hypochromic anemia, thrombocytopenia, increase ESR and 24 urine collection showed increase in total hydroxyproline: 111.0 mg/day (N: 15.0 - 42.0 mg/day) and free hydroxyproline: 1.3mg/dl (N: < 0.4 mg/dl). Pelvi-abdominal ultrasonography revealed mild splenomegaly. A four mm punch biopsy specimen was obtained from the border of the ulcer including the adjacent intact elevated margin. Hematoxylin and eosin (H&E) stained sections revealed non- specific inflammatory infiltrate in the dermis with thin collagen fibers and perivascular amorphous material
(Fig 2a) which stained +ve with Congo red stain (Fig 2b).
 | Fig
2a: hin collagen fibers with perivascular amorphous material (
H&Ex100). |
|
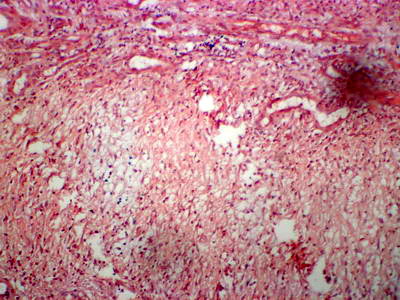 | Fig
2b: Amorphous vascular deposits with congo red +ve (Congo
red x120). |
|
The clinical, histopatholgical and laboratory findings were in keeping with the diagnosis of prolidase deficiency syndrome. The patient was treated with topical and systemic steroids, dapsone, high doses of vitamin C and packed RBCS transfusion every month for 2 months. He got satisfactory improvement with healing of the ulcers with scarring. Discussion
Prolidase deficiency (PD) is a rare, autosomal recessive inborn error of collagen metabolism [1,2] first described by Goodman in 1968. It is caused by a defect in the PEPD gene on chromosome 19.[3,4] In addition to its clinical features, some patients with PD develop systemic lupus erythematosus later in life, suggesting that PD may also be a risk factor for this disease.[5,6] Collagen is degraded to iminodipeptides, which are then broken down into amino acids that can be resynthesized to form collagen. [7] One of the enzymes that degrade iminodipeptides is prolidase. Specifically, it cleaves dipeptides with proline or hydroxyproline at the C-terminus end. [1,8] Decreased activity of prolidase causes a concomitant increase in urinary excretion of dipeptides (iminodipeptiduria). This iminodipeptiduria represents a deficient recycling of proline and leads to impaired collagen synthesis and wound healing.[1,9,10,11]In patients with PD, amyloid deposits have been found within the vessels of the skin and internal organs, including the spleen. Splenic amyloid deposits lead to splenomegaly and subsequent splenic dysfunction. This may be the cause of the characteristic increased risk of infection in patients with PD [6]. Neutrophils have also been found to play a role in clinical manifestations of PD. Increased iminodipeptide levels lead to proliferation of neutrophils, which, in turn, generate superoxide molecules that mediate inflammation and tissue destruction. [12] In addition, neutrophils release collagenases and, along with histamine from mast cells, mediate further tissue inflammation and subsequent tissue destruction. [13] Prolidase deficiency syndrome characterized by various signs and symptoms have been reported in the literature
(Table 1). [14]
|
Dermatologic
• Chronic lower extremity ulcerations
•Scarring
• Telangiectasias
• Purpura
• Lymphedema
•Poliosis
• Photosensitivity
•Pruritus
• Hyperkeratosis
•Low hairline
• Simian crease
•Saddle nose
• Premature graying of the hair
•Hypohidrosis
•Hypertrichosis
•Hypoplastic jaw •Curved finger
nail
• Frontal bossing
• Beaklike nose
• Thick lips
• Micrognathia
• Hirsutism
•Protrusion of mandible •
Hypertelorism
•Gynecomastia
•Hypogonadism
• Scaly erythematous maculopapular rash
•Thinning of skin over the abdomen, with prominent
veins
• Thickening of skin of lower extremities
• Pustules on face, back, and extremities
• Long eyebrows and eyelashes
Central nervous system
• Mental handicap
Lymphoreticular
• Splenomegaly
•Hepatomegaly
Immune system
•Frequent infections, especially upper respiratory
infection and otitis media
Otorhinolaryngologic
•
Deafness •Arched
palate
•Purulent tonsillitis
•Dental caries
•Abnormal middle ear
•Sinusitis
•Perforated nasal septum
•Hypoplasia of enamel
Ocular
• Keratitis
• Corneal ulcers
•
Myopia • Bilateral
ptosis
• Retinal pigmentary changes
Musculoskeletal
• Hyperextensible joints •
Osteoporosis
• Hand muscle wasting •
Short stature
• Short Achilles tendon •
Dislocated hip
• Foot anomalies
•Arachnodactyly
• Coxa valga
• Genu valgum
•Waddling gait with external rotation of femur
Laboratory abnormalities
•Iron-deficiency anemia
•Thrombocytopenia
• Hypergammaglobulinemia •
Aminoaciduria |
Table 1: Signs and
symptoms of prolidase deficiency PD can be diagnosed based on decreased prolidase activity in the blood and increased levels of urinary iminodipeptides. [1] Prolidase is a ubiquitous enzyme; however, it exerts its greatest activity in the red blood cells and kidney.[8] To determine the prolidase level, an enzyme assay of prolidase can be done using red blood cells and fibroblasts.[1,8] To establish the presence of iminodipeptiduria, the patient's urine may be analyzed using urine electrophoresis on paper or thin-layer cellulose acetate and column chromatography; direct chemical ionization mass spectrometry can also detect iminodipeptiduria.[2,5,8] In addition, PD can be diagnosed in utero by using chorionic villous sampling or amniocentesis.[5] Laboratory abnormalities associated with PD include iron-deficiency anemia, thrombocytopenia, and hypergammaglobulinemia.[9,16] However, these are neither diagnostic nor pathognomonic for PD. Skin histopathology is almost normal in patients with PD[2], although there may be a decrease in the size of collagen fibers or fragmentation of collagen fibers with impaired aggregation. Elastic fibers are normal,[15] but dense bodies have been found in capillary endothelium.[16] Angiopathy, perivascular neutrophilic infiltrate, dermal sclerosis, occlusion of capillary and arteriole walls with thrombi, and fibrosis of vessels walls have been demonstrated in patients with PD.[16,17] Congo red staining has confirmed the presence of amyloid, which can occlude the vessels.[6,12,16] Although our patient had typical features of prolidase deficiency, the differential diagnosis included Wegener granulomatosis (WG), which has cutaneous involvement as the presenting sign in 9-14 percent of cases. WG is a multisystem disease with a predilection for the lung, kidney, and eye [18]. It can present with a limited cutaneous form in which there is no evidence of systemic involvement and negative ANCA testing [19]. The presence of histopathologic findings showing palisading granulomatous inflammation with or without granulomatous vasculitis, and formation of neutrophilic microabscesses with a diffuse c-ANCA value is considered diagnostic of WG [20]. In our case, ANCA testing was repeatedly negative, there was no subsequent development of systemic disease, and no systemic involvement was noted during a 12 month follow up; the possibility of WG was excluded by negative ANCA testing together with clinicopathologic findings suggestive of prolidase deficiency. Various topical, systemic, and surgical treatments for leg ulcers secondary to PD have been reported in the literature; however, none has been shown to be effective in randomized, controlled trials. Topical antibiotics, such as gentamicin, polymyxin, and colistin, are suggested to be effective in controlling infection.[7] When infection develops, oral or intravenous antibiotics become necessary.[16] Topical 5% proline and 5% glycine combined in an ointment base have been shown to be more effective in healing ulcers from PD than topical 5% proline in ointment alone.[10] In addition, a combination of topical and systemic growth hormone healed recalcitrant leg ulcers in 1 patient.[4] Various systemic therapies that have shown efficacy in a handful of patients with leg ulcers from PD include a combination of vitamin C and manganese, with or without proline; a combination of zinc sulfate and vitamin C; and growth hormone.[1,6,7,9,10,21] Periodic red blood cell transfusions and apheresis erythrocyte exchanges have also proven to be effective in healing chronic leg ulcers.[6,22] Dapsone was used to heal patient's recalcitrant leg ulcers.[16] Surgical treatment options for ulcers secondary to PD include autologous split-thickness skin grafts or limb amputations. [9,16] We suggest that patients who develop chronic and recurrent leg ulcers, even those that resemble other inflammatory disorders, should be examined for prolidase deficiency. References
1. 1. Berardesca E, Fideli D, Bellosta M, et al. Blood transfusions in the therapy of a case of prolidase deficiency. Br J Dermatol. 1992;126:193-195.
2. Der Kaloustian VM, Freij BJ, Kurban AK. Prolidase deficiency: an inborn error of metabolism with major dermatological manifestations. Dermatologica. 1982;164:293-304.
3. Endo F, Matsuda I. Molecular basis of prolidase (peptidase D) deficiency. Mol Biol Med. 1991;8:117-127.
4. Monafo V, Marseglia GL, Maghnie M, Dyne KM, Cetta G. Transient beneficial effect of GH replacement therapy and topical GH application on skin ulcers in a boy with prolidase deficiency. Pediatr Dermatol. 2000;17:227-230.
5. Jaeken J: Prolidase deficiency. Orphanet Encyclopedia January 2004. Available at:http://www.orpha.net; accessed September 20, 2004;.
6. Bissonnette R, Friedmann D, Giroux JM, et al. Prolidase deficiency: a multisystemic hereditary disorder. J Am Acad Dermatol. 1993;29:818-821.
7. Arata J, Hatakenaka K, Oono T. Effect of topical application of glycine and proline on recalcitrant leg ulcers of prolidase deficiency. Arch Dermatol. 1986;122:626-627.
8. Freij BJ, Der Kaloustian VM. Prolidase deficiency. A metabolic disorder presenting with dermatologic signs. Int J Dermatol. 1986;25:431-433.
9. Milligan A, Graham-Brown RA, Burns DA, Anderson I. Prolidase deficiency: a case report and literature review. Br J Dermatol. 1989;121:405-409.
10. Jemec GB, Moe AT. Topical treatment of skin ulcers in prolidase deficiency. Pediatr Dermatol. 1996;13:58-60.
11. Arata J, Umemura S, Yamamoto Y, Hagiyama M, Nohara N. Prolidase deficiency: its dermatological manifestations and some additional biochemical studies. Arch Dermatol. 1979;115:62-67.
12. Yasuda K, Ogata K, Kariya K, et al. Corticosteroid treatment of prolidase deficiency skin lesions by inhibiting iminodipeptide-primed neutrophil superoxide generation. Br J Dermatol. 1999;141:846-851.
13. Kokturk A, Kaya TI, Ikizoglu G, Koca A. Prolidase deficiency. Int J Dermatol. 2002;41:45-48.
14. Trent JT, Kirsner RS. Leg ulcers secondary to prolidase deficiency. Adv Skin Wound Care. 2004;17:468-472.
15. Leoni A, Cetta G, Tenni R, et al. Prolidase deficiency in two siblings with chronic leg ulcerations. Clinical, biochemical, and morphologic aspects. Arch Dermatol. 1987;123:493-499.
16. Ogata A, Tanaka S, Tomoda T, et al. Autosomal recessive prolidase deficiency. Three patients with recalcitrant ulcers. Arch Dermatol. 1981;117:689-697.
17. Arata J, Tada J, Yamada T, et al. Angiopathic pathogenesis of clinical manifestations in prolidase deficiency. Arch Dermatol. 1991;127:124-125.
18. Kavala M, Zindanci I, Sudogan S, Turkoglu Z, Sarigul S. Ulcus cruris associated with prolidase deficiency. Dermatol Online J. 2006;12:24.
19. Gibson LE, Specks U, Homburger H. Clinical utility of ANCA tests for the dermatologist. Int J Dermatol. 2003;42:859-869.
20. Kradin RL, Mark EJ. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 18-2002. A 48-year-old man with a cough and bloody sputum. N Engl J Med. 2002;346:1892-1899.
21. Larreque M, Charpentier C, Laidet B, et al. [Prolidase and manganese deficiency. Apropos of a case: diagnosis and treatment]. Ann Dermatol Venereol. 1982;109:667-678.
22. Lupi A, Casado B, Soli M, et al. Therapeutic apheresis exchange in two patients with prolidase deficiency. Br J Dermatol. 2002;147:1237-1240.© 2011 Egyptian Dermatology Online Journal |

