|
|
Abstract
Primary systemic amyloidosis is a rare disorder
characterized by multi-systemic homogenous hyaline amyloid material. We
report a case of 68 years old male presented with generalized weakness,
fatigue, anorexia and impaired taste sensation. Muco-cutaneous lesions like,
macroglossia with bullous haemorrhagic lesions, peri-orbital purpura, and
nail changes were present. He had no cardiomegaly, hepatomegaly,
splenomegaly or renal involvement. His skiagram chest showed minimal left
sided pleural effusion which had no pus cells despite high protein content.
The diagnosis was established on the basis of classical cutaneous lesions.
Diagnosis was confirmed by tongue biopsy using Haematoxylin and Eosin
staining and crystal violet staining. Polarized microscopy was not done
because of unavailability. We report this case because of the sheer rare
presentation and diagnosis is considered in dermatologist's differentials.
This case report suggests that skin lesions may provide a clue for an early
diagnosis of systemic amyloidosis, which lengthens the survival period of
the patient.
Introduction
Primary systemic amyloidosis is a plasma cell
dyscrasia of unknown cause. The systemic form is sub-classified as primary,
secondary, familial and amyloidosis associated with multiple myeloma [1].
The accepted nomenclature is AX, where A represents amyloidosis and X
indicates type of protein in the fibrils, AL is for light chains, AA for
secondary amyloidosis (in chronic infectious and inflammatory diseases)
while AF indicates familial amyloidosis. Systemic amyloidosis affects both
sexes with male preponderance; onset usually begins in the sixth decade. The
kidneys are most frequently affected organ (80%), followed by the heart
(40%). Carpel tunnel syndrome is present in 25% of cases, sensory neuropathy
(18%), postural hypotension (16%) are the other common manifestations.
Specific mucocutaneous lesions occur in 29% to 40% of cases which gives an
early indication of the existence of an underlying plasma cell dyscrasia [2,3].
No satisfactory treatment for systemic amyloidosis has been discovered. In
this article, we report this case because of rare presentation, without
plasma cell dyscrasia and no renal, cardiac or hepatic involvement. Presence
of macroglossia and typical mucocutaneous lesions help the dermatologist to
reach the diagnosis at an early stage and when the initiation of therapy
starts before the onset of organ failure, survival may be prolonged.
Case Report
A 68 year old male presented in medical outpatients
department of Era's Lucknow medical college hospital with complaints of
anorexia, generalized weakness, impaired taste sensation and progressive
weight loss of approximately 10 kg in the last year. There was no other
systemic complaint or history of any other chronic illness in the past.
Family history was insignificant. On examination he had normal vital
parameters and blood pressure was 140/70 mmHg with no postural hypotension.
There was mild pallor and pedal oedema. The systemic examination did not
reveal any abnormality. In view of presence of peri-orbital pigmentation and
lesions present in oral cavity he was referred to seek dermatologist
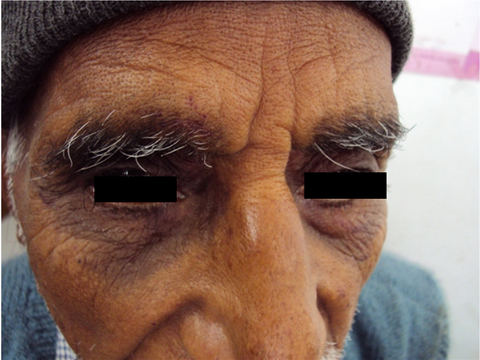
consultation. On dermatological examination peri-orbital pigmentation was
found to be purpuric lesion around the eyelids (fig.
1). Ecchymotic lesions were also present over
the lips, and on buccal mucosa (fig. 2).
There was ecchymoses at the site of venipuncture. The tongue was diffusely
enlarged, firm, fissured and haemorrhagic bulla was present on the surface.
Teeth indentation marks were present around its lateral border. Multiple
shiny smooth, firm yellowish papules up to 0.5cm in size were present on the
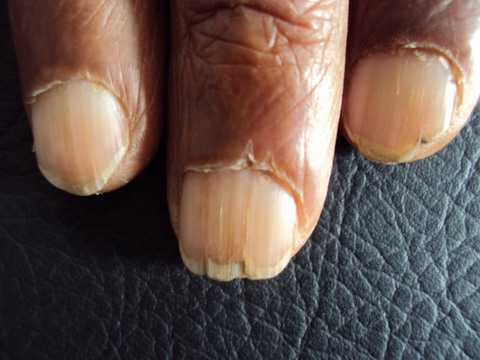
retro-auricular area and over eyelids. Nail changes were present,
longitudinal striations, crumbling, brittleness, ragged cuticle and partial
onycholysis were present (fig. 3).
On investigations, Hb was 8gm%, ESR was 56 mm at first
hour. Urine showed albumin1+ with no Bence Jones proteins. Serum and urine
electrophoresis was normal. All other hematological and biochemical reports
including coagulation profile were normal. The x-ray chest showed minimal
pleural effusion on left side which when aspirated showed protein content
3.5gm/dl with no pus cells and was sterile on culture. RA factor, ANA and
anti ds DNA were negative. Bone marrow examination was also normal. ECG and
2D Echo were normal. Abdominal ultrasound did not reveal any abnormality. No
pathology was demonstrated in bone survey.
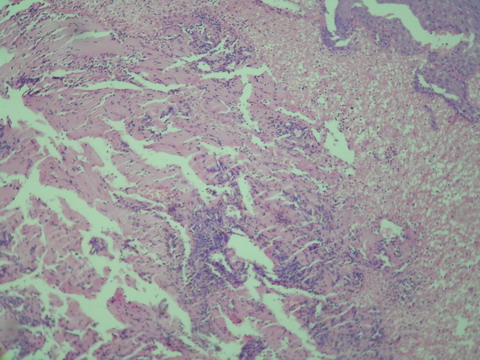
The histopathological examination of the biopsy taken
from the lesion on the tongue revealed homogenous eosinophilic amorphous
material and infiltration with lymphocytes seen on haematoxylin and eosin
staining (fig. 4)
and revealed metachromatic staining on crystal violet. Polarized microscopy
was not done because of unavailability.
Discussion
Amyloidoses are protein confirmation disorders, in
which different soluble proteins aggregate as extracellular insoluble
fibrils. In immunoglobulin light chain amyloidosis (AL), monoclonal light
chains undergo conformational changes leading to their aggregation into
amyloid fibrils that can deposit in virtually every organ, with the
exception of parenchymal brain tissue [4].
This process causes organ dysfunction and, if it is not halted by therapy,
it leads to patient's death. Median survival of primary systemic amyloidosis
without myeloma is up to 14.7 months. Unusual longevity for 19 years have
been reported with combined melphalan, prednisolone and colchicine treatment
and is the longest reported in literature [5].
Non specific presentation delays 1-2 years in reaching the diagnosis.
In our case, the presenting symptoms are fatigue,
weight loss, breathlessness, reduced taste perception and mucocutaneous
presentations. Impaired taste perception is a frequently unnoticed sensory
impairment in primary systemic amyloidosis [6].
Clinically evident mucocutaneous involvement in
primary systemic amyloidosis is an early pointer. Oral manifestations have
been reported in 39% of patients with amyloidosis [7].
Amyloidosis deposition in the tongue of multiple myeloma occurs frequently
and can result in macroglossia, which is the most common oral finding [7,8,9].
An enlarged tongue resulting in apertognathia and tooth indentations along
the lateral border can be the first clinical sign of amyloidosis [10,11,12].
In our case, tongue of the patient is enlarged, firm,
fissured and hemorrhagic bulla were present on its surface. Teeth
indentations were present along the lateral border of tongue.
Amyloid infiltration of vessel wall causes capillary
fragility, which bleeds on minor trauma or even spontaneously. Purpura,
ecchymoses and petechiae occur commonly in the skin and mucous membranes due
to intracutaneous haemorrhage. In our case, peri-orbital purpura and
ecchymotic lesions were present on lips and on buccal mucosa. Ecchymoses was
present at the site of veni-puncture. Purpuric lesions with normal platelet
count and normal coagulation profile should suggest the possibility of
capillary fragility. Senile purpura which also comes in differentials, is
also due to capillary fragility but is commonly seen on extremities and
rarely involve the peri-orbital and over the lips and buccal mucosa.
Nail dystrophy is not unusual in systemic amyloidosis.
In our case longitudinal ridging, ragged cuticles and splitting of nails was
present. There are reported instances of nail dystrophy seeing the only
feature at presentation [13,14].
Biopsy is the definitive investigation for
amyloidosis. Fine-needle fat-pad biopsy and rectal biopsy have both been
hailed as the investigation of choice, each giving positive results in 80-90
percent of cases [15,16,17].
Haematoxylin and eosin staining suggests the possibility of amyloidosis but
Congo red staining confirms the diagnosis. Skin biopsy characteristically
shows diffuse amyloid deposition in the form of nodules and plaques. There
may be amyloid infiltration of blood-vessel walls, around individual fat
cells, and in pilo-sebaceous units.
The clinical diagnosis was confirmed by tongue biopsy
on H & E stain, the specimen revealed homogenous eosinophilic aggregates and
infiltration with lymphocytes and metachromatic stain with crystal violet.
The prognosis for AL amyloidosis is poor, with death
usually resulting from cardiac or renal failure. The mean survival from
diagnosis is approximately 15 months [7].
Treatment is aimed at decreasing amyloid production and deposition and
promoting lysis of deposits. Current treatment options include oral
melphalan with or without prednisone, as well as stem-cell harvest and
transfer, with intravenous melphalan or other chemotherapeutic agents.
Supportive measures include symptomatic treatment for organ impairment, such
as diuretics for cardiac impairment, or hemodialysis in the case of renal
failure. Stem-cell harvest and transfer, after the use of either melphalan
or other chemotherapeutic agents, has also been shown to improve survival. [18]
The case of systemic amyloidosis is presented because
of its rare occurrence and in this case no other organ was involved and only
mucocutaneous findings help in reaching the diagnosis which was confirmed by
tongue biopsy. Clinically evident mucocutaneous involvement in primary
systemic amyloidosis is an early pointer and this condition is considered in
the dermatologist's differential. So, early diagnosis and initiation of
therapy before the onset of organ failure is essential to lengthen the
survival period.
References
1. Lechmann HJ, Hawkins PN. Amyloidosis
and the Skin. In: Wolff K, Goldsmith LA, Kat SI, Gilchrest BA, Paller AS,
Leffell DJ, editors. Fitzpatricks dermatology in General Medicine. 7th ed.
New York: Mc Graw Hill; p. 1256-65, 2008
2. Brathnach
SM. Amyloid and amyloidosis. J Am Acad Dermatol 1988; 18: 1-.16
3. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and
possible association with systemic amyloidosis. Int J Dermatol 2002; 41:
127- 132
4. Merlini G, Bellotti V. Molecular
mechanisms of amyloidosis. N Engl J Med 2003; 349: 583- 596
5. Fritz DA, Luggen ME and Hess EV. Unusual longevity in primary systemic
amyloidosis. Am J Med 1989; 86: 24
6. Marinone MG,
Marinone MG, Merlini G. Reduced taste perception in AL amyloidosis. A
frequently unnoticed sensory impairment. Haematologica 1996; 81: 110- 115
7. Kyle RA, Griepp PR. Amyloidosis (AL): Clinical and laboratory features in
229 cases. Mayo Clin Proc 1983; 58: 665- 683
8.
Jacobs P, Sellars S, King HS. Massive macroglossia, amyloidosis and myeloma.
Postgrad Med J 1988; 64: 696- 698
9. Raubenheimer EJ,
Dauth J, de Coning JP. Multiple myeloma presenting with extensive oral and
perioral amyloidosis. Oral Surg Oral Med Oral Pathol 1986; 61: 492- 497
10. Loh FC, Ravindranathan N, Yeo TF. Amyloidosis with oral involvement:
Case report. Aust Dent J 1990; 35: 14- 18
11. Smith
A, Speculand B. Amyloidosis with oral involvement. Br J Oral Maxillofac Surg
1985; 23: 435- 444
12. Babajews A. Occult multiple
myeloma assosciated with multiple myeloma of tongue. Br J Oral Maxillofac
Surg 1985; 23: 298-303
13. Fanti PA, Tosti A,
Morelli R, Galbiati G. Nail changes as the first sign of systemic
amyloidosis. Dermatologica 1991; 183(1): 44- 46
14.
Wheeler GE, Barrows GH. Alopecia Universalis: A manifestation of occult
amyloidosis amyloidosis and multiple myeloma. Arch Dermatol 1981; 117(12):
815
15. Duston MA, Skinner M, Shirahama T, Cohen AS.
Diagnosis of amyloidosis by abdominal fat pad aspiration: Analysis of four
years' experience. Am J Med Mar 1987; 82(3): 412- 414
16. Libbey CA, Skinner M, Cohen AS. Use of abdominal fat pad aspirate in the
diagnosis of systemic amyloidosis. Arch Int Med 1983; 143(8): 1549- 1552
17. Blum A, Sohar E. The diagnosis of amyloidosis: Ancillary procedures.
Lancet 1962; 1(7232): 721- 723
18. Comenzo RL,
Vosburgh E, Falk RH, Sanchorawala V et al. Dose-intensive melphalan with
blood stem-cell support for the treatment of AL (amyloid light chain)
amyloidosis: survival and responses in 25 patients. Blood 1998; 91(10):
3662- 3670© 2010 Egyptian
Dermatology Online Journal |